


LIPOSOME BASED SUSTAINED RELEASE DELIVERY SYSTEM FOR ANTICANCER DRUG
HTML Full TextLIPOSOME BASED SUSTAINED RELEASE DELIVERY SYSTEM FOR ANTICANCER DRUG
Kriti Pateriya * 1, Swatantra K. Kushwaha 1, Rohit Kumar Bijauliya 2 and Dilip Kumar Chanchal 3
Paramveer Singh Institute of Technology (PSIT) 1, Kanpur - 209305, Uttar Pradesh, India.
Department of Pharmacognosy 2, Bundelkhand University, Jhansi - 284128, Uttar Pradesh, India.
Society of Pharmaceutical Sciences and Research 3, Panchkula - 134112, Haryana, India.
ABSTRACT: Most of the existing anticancer drugs are very potent small molecules; their efficacy is constrained by their systemic toxicity, narrow therapeutic window, low drug loading, size control, scale up, cost of formulation but also as a result of drug resistance and limited cellular entry. Due to these obstacles, controlled and targeting or localized release technology has been replacing the systemic administration and has shown lots of potential for cancer treatment. Liposomes can be used to provide a sustained release of drugs, which require a prolonged plasma concentration at therapeutic levels to achieve the optimum therapeutic efficacy. In the present work, we formulate liposome containing the anticancer drug ‘6 Mercaptopurine’ for sustained delivery. The drug incorporation was carried out using the ether injection method. The cumulative drug release for the formulations, prepared by ether injection method, F1, F2, F3, F4, F5, and F6 were found to be 66.33%, 63.5%, 68.78%, 62.62%, 63.8%, and 61.25% respectively. The drug release kinetics studies suggest that in the formulations prepared by ether injection method, the best fit model was first order for formulation F1, while for formulations F2, F3, F4, and F6 the best fit model was Korsmeyer and for formulation F5 the best fit model was zero and hixon. ‘n’ exponent value for Pappas model, for formulations F1, F2, F3, F4, F5, and F6 is greater than 0.89 indicating that formulation is released by super case 2 transport mechanism.
Keywords: Liposomes, Drug delivery vehicles, Anticancer drug
INTRODUCTION: Cancer is amongst the top three killers in modern society, next to the heart and cerebrovascular diseases. Treating cancer has always been a challenge because cancer chemotherapeutic agents are cytotoxic and cannot differentiate cancer cells from normal cells.
This leads to the destruction or impairment of vital organs particularly those that have a high rate of cell division like the liver, GI lining, hair, and skin; in addition to the killing off the cancer cells if their bio-distribution is not properly controlled and the therapeutic agents not targeted towards the cancer cells or tissues.
Thus targeting continues to be the Holy Grail in anticancer therapy. Liposomes were first produced in England in the ’60s, by Bangham who was studying phospholipids and blood clotting 1. Liposomes are spherical vesicles with concentric phospholipid bilayers 2 that are formed spontaneously in aqueous solution 3. The word liposome comes from two Greek words, lipos (fat) and soma (body or structure) 4, 5. Lipid bilayered membrane encloses an aqueous core, and hydrophilic drugs may get entrapped in the central aqueous core of the vesicles while lipophilic drugs are entrapped within the bilayered membrane 6.
Liposomes are extensively used as carriers for numerous molecules in the cosmetic and pharmaceutical industries. Liposomes were introduced as drug delivery vehicles in the 1970s 7. It has been displayed that phospholipids impulsively form closed structures when they are hydrated in aqueous solutions. Such vesicles which have one or more phospholipid bilayer membranes can transport aqueous or lipid drugs, depending on the nature of those drugs 8. Generally, liposomes are definite as spherical vesicles with particle sizes ranging from 30 nm to several micrometers 9. The history of liposomes can be divided into three periods: Genesis (1968-75), middle age (1975 - 85) and modern era (1985 onwards) 10.
These ‘liposomes’ form a barrier around their contents, which is resistant to enzymes in the mouth and stomach, alkaline solutions, digestive juices, bile salts, and intestinal flora that are generated in the human body, as well as free radicals. The contents of the liposomes are, therefore, protected from oxidation and degradation. This protective phospholipid shield or barrier remains undamaged until the contents of the liposome are delivered to the exact target gland, organ, or system where the contents will be utilized11.
Many researchers have studied and worked on liposomes, but small numbers of liposomal products have been approved to be used in human. This may be due to many reasons such as High cost of liposome production especially in large scales, the toxicity of some liposomal formulations, relative short half-life, instability, low solubility, low entrapment of molecules and compounds into vesicles and sometimes phospholipid undergoes oxidation and hydrolysis 12, 13, 14. When phospholipids are placed in water, and sufficient energy is provided from sonication, heating, homogenization, etc., results in the arrangement of the lipids and formation of bilayer vesicles 15. The phenomenon can be a result of the critical micelle concentration (CMC) of phospholipids in water. The CMC may be defined as the concentration of lipid in water, above which the lipid forms micelles or bilayer structures rather than remaining in solution as monomers.
MATERIALS AND METHODS:
6-Mercaptopurine: 6-Mercaptopurine (6, 7-dihydro-3H-purine-6-thione) is an antimetabolite anti-neoplastic agent with immunosuppressant properties. It interferes with nucleic acid synthesis by inhibiting purine metabolism and is used, usually in combination with other drugs, in the treatment of or in remission maintenance programs for leukemia and the treatment of Paediatric non-Hodgkin's lymphoma.
Phospholipids: Phospholipids are the major structural components of biological membranes. The most common phospholipid used in liposomal preparation is phosphatidylcholine (PC). Phosphatidylcholine is an amphipathic molecule containing 6, 7 Molecules of PC are not soluble in water.
Cholesterol: Cholesterol dose not by itself form bilayer structure, but can be incorporated into phospholipid membranes in very high concentration up to 1:1 or even 2:1 molar ratio of cholesterol to phosphatidylcholine. Cholesterol inserts into the membrane with its hydroxyl group oriented towards the aqueous surface and aliphatic chain alignment parallel to the acyl chains in the center of the bilayer. The high solubility of cholesterol in phospholipid liposome has been attributed to both hydrophobic and specific head group interaction, but there is no unequivocal evidence for the arrangement of cholesterol in the bilayer.
Methods:
Selection of the Best Method for Liposome Preparation: Ether injection (solvent vaporization) A solution of lipids dissolved in diethyl ether or ether methanol mixture is gradually injected to an aqueous solution of the material to be encapsulated at 55 °C to 65 °C or under reduced pressure. The consequent removal of ether under vacuum leads to the creation of liposomes. An advantage of the ether injection method compared to the ethanol injection method is the removal of the solvent from the product, enabling the process to be run for extended periods forming a concentrated liposomal product with high entrapment efficiencies.
Preparation of Drug Loaded Liposome: The required amount of phospholipid and cholesterol were dissolved in ether, and the lipophilic drug was added to the organic one. The resulting organic phase was injected using a syringe pump in a defined volume of distilled water under magnetic stirring at a temperature of 55-65 °C under reduced pressure. The ether vaporizes upon contacting the aqueous phase, and the dispersed lipid forms primarily unilamellar liposomes. 40 ml formulation was prepared.
Evaluation: Prepared formulations were evaluated by the following tests:
- Entrapment
- In-vitro release studies
- Drug Release Kinetics studies Sterility
Drug Entrapment: All the formulation were subjected for determination of drug entrapment. 10 ml of liposome formulation was pipette out and was transferred to a 100 ml volumetric flask containing 20 ml of 0.1N NaOH solution then sonicated and filtered through Whatman filter paper. The filtrate was finally diluted with 0.1N NaOH, 6 mercaptopurine concentration was then determined at 307 nm by using UV-Vis spectrophotometer.
In-vitro Release Studies: In-vitro release studies were performed using a dialysis membrane method. Phosphate buffer pH 7.4 (100 ml) was placed in a 250 ml beakers. The beaker was assembled on a magnetic stirrer, and the medium was equilibrated at 37 ± 0.5 ºC. Dialysis membrane was taken, and one end of the membrane was sealed. Liposome formulation was filled in the dialysis membrane, and another end was closed.
The dialysis membrane containing the sample was suspended in the medium. Aliquots were withdrawn (5 ml) at specific intervals, filtered and the medium was immediately replaced with the same quantity of fresh buffer solution. The Aliquots was measured for the amount of the drug by using UV spectrophotometer at 307 nm.
Drug Release Kinetics: The drug release data obtained for each formulation were fitted into the mathematical model given below to determine the drug release kinetics of the prepared formulation:
- Cumulative drug release v/s Time (Zero Order kinetics)
- Log percent drug remaining to be released v/s Time (First-order rate kinetics)
- Cumulative percent drug released v/s Root time (Higuchi matrix)
- (Cube root of % drug remaining to be released)1/3 v/s Time (Hixson-Crowell erosion equation)
To understand the mechanism of drug release, the data of drug release was fitted in the Korsmeyer-Peppas model. Where Log of cumulative percent drug released was plotted against the Log time. The model was used to understand the mechanism of drug release by analyzing ‘n’ as the diffusion exponent. According to this, the value of ‘n’ below 0.45 then Fickian mechanism governs the drug release whereas if the value of ‘n’ is between 0.45- 0.85 than Non- Fickian mechanism takes place. If the value of ‘n’ is 0.85 or exceeds the 0.85 than release mechanism is governed by the Case II transport or super Case II transport respectively.
RESULTS AND DISCUSSION:
Selection of the Best Method for Liposome Preparation: Ether injection (solvent vaporization) A solution of lipids dissolved in diethyl ether or ether methanol mixture is gradually injected to an aqueous solution of the material to be encapsulated at 55 °C to 65 °C or under reduced pressure. The consequent removal of ether under vacuum leads to the creation of liposomes. An advantage of the ether injection method compared to the ethanol injection method is the removal of the solvent from the product, enabling the process to be run for extended periods forming a concentrated liposomal product with high entrapment efficiencies.
Incorporation of Drug in Liposome: The required amount of phospholipid and cholesterol were dissolved in ether, and the lipophilic drug was added to the organic one. The resulting organic phase was injected using a syringe pump in a defined volume of distilled water under magnetic stirring at a temperature of 55-65 °C under reduced pressure. The ether vaporizes upon contacting the aqueous phase, and the dispersed lipid forms primarily unilamellar liposomes. 40 ml formulation was prepared in Table 1.
TABLE 1: FORMULATION DESIGN FOR PREPARATION OF DRUG LOADED LIPOSOME BY ETHER INJECTION METHOD USING DIETHYL ETHER AS A SOLVENT
| Formulation | Lipid | Cholesterol | Drug | Water |
| F1 | 5 (200 mg) | 1 (40 mg) | 100 mg. | 40 ml |
| F2 | 6 (480 mg) | 2 (80 mg) | 100 mg. | 40 ml |
| F3 | 7 (840 mg) | 3 (120 mg) | 100 mg. | 40 ml |
| F4 | 4 (160 mg) | 1 (40 mg) | 100 mg. | 40 ml |
| F5 | 3 (240 mg) | 2 (80 mg) | 100 mg. | 40 ml |
| F6 | 2 (240 mg) | 3 (120 mg) | 100 mg. | 40 ml |
Evaluation:
Drug Entrapment: Table 2 and Fig. 1 shows the percent entrapment of drug for the formulations prepared by ether injection method. Entrapment for the formulations prepared by ether injection method F1, F2, F3, F4, and F5 was found to be 48%, 64%, 72%, 64%, 40% and 32% respectively.
TABLE 2: PERCENTAGE DRUG ENTRAPMENT OF FORMULATIONS PREPARED BY ETHER INJECTION METHOD
| Formulation | % Drug Entrapment |
| F1 | 48% |
| F2 | 64% |
| F3 | 72% |
| F4 | 64% |
| F5 | 40% |
| F6 | 32% |
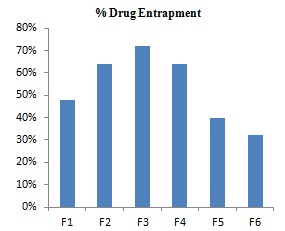
FIG. 1: BAR CHART SHOWING PERCENT ENTRAPMENT FOR PREPARED FORMULATIONS
In-vitro Release Studies: The release profile for the formulation predicts how a delivery system might function and gives valuable insight into its in-vivo behavior. The various formulations of 6 Mercaptopurine were subjected to in-vitro release studies. These in-vitro release studies were carried out using 0.1 N NaOH as the dissolution medium. The Cumulative drug release data concerning time for the various formulations are shown in Table 3, the release pattern concerning time for the formulations are shown in Fig. 2 and 3. The cumulative drug release for the formulations, prepared by ether injection method F1, F2, F 3, F4, F5, and F6 were found to be 66.33%, 63.5%, 68.78%, 62.62%, 63.8%, and 61.25% respectively.
The formulation F2 and F4 showed quite a similar pattern of release barring the first 5 h. In this duration formulation, F1 showed a slight burst release in the Ist h followed by a linear pattern of release while F2 showed a slow initial release, in the Ist h, followed by a linear pattern, in the entire period of 3 h F4 showed significantly less drug release as compared to other formulation. On the other hand showed a slow initial release in the first 90 min, followed by burst release till the 4th h and this was followed by a linear release pattern. The formulation of F2 and F4 showed an almost linear pattern of release. Formulation F6 also showed an almost linear pattern of release except for the period between 3rd to 5th h in which it showed a slight burst release. Overall these 6 formulations prepared by the ether injection method released lesser drug content within 6 h and thus were found to be more suitable for sustained release, especially formulation F6.
Drug Release Kinetics: Plots of zero order, first order, Higuchi matrix, Pappas and Hixson Crowell for the formulations were plotted. The regression coefficient (r2) values of zero order, first order, Higuchi matrix, Hixson-Crowell, Korsmeyer Pappas and the ‘n’ values for Korsmeyer Pappas are tabulated in Table 4 for the formulations prepared by ether injection method respectively.
From the Table 4, it is clear that in case of formulations prepared by ether injection method, the best fit model was first order for formulation F 1, while for formulations F2, F3, F4, and F6 the best fit model was Korsmeyer and for formulation F 5 the best fit model was zero and Hixon. ‘n’ exponent value for Pappas model, for formulations F1, F2, F3, F4, F5, and F6 is greater than 0.89 indicating that formulation is released by super case II transport mechanism.
TABLE 3: IN-VITRO DRUG RELEASE OF 6-MERCAPTOPURINE FROM FORMULATIONS PREPARED BY ETHER INJECTION METHOD
| Time
(h) |
Cumulative % Drug Release | |||||
| F1 | F2 | F3 | F4 | F5 | F6 | |
| 0.5 | 3.33 ± 0.12 | 0.37 ± 0.33 | 1.00 ± 0.12 | 0.75 ± 0.73 | 1.8 ± 0.08 | 3.00 ± 1.54 |
| 1 | 8.83 ± 0.73 | 1.5 ± 0.76 | 3.78 ± 0.09 | 2.625 ± 1.54 | 6.8 ± 0.07 | 5.75 ± 1.26 |
| 1.5 | 16.83 ± 1.54 | 5.625 ± 1.23 | 7.44 ± 1.12 | 5.675 ± 1.36 | 11.28 ± 1.20 | 11.25 ± 1.34 |
| 2 | 22.17 ± 2.04 | 8.375 ± 1.32 | 11.44± 2.12 | 7.51 ± 1.54 | 17.94 ± 2.34 | 15.75 ± 2.36 |
| 2.5 | 29.67 ± 2.12 | 11.75 ± 1.97 | 16.67 ± 2.08 | 13.62 ± 2.11 | 26.8 ± 0.25 | 19.00 ± 2.31 |
| 3 | 33.83 ± 2.32 | 16.25 ± 2.81 | 22.56 ± 2.43 | 18.12 ± 0.56 | 31.2 ± 0.46 | 24.50 ± 1.35 |
| 3.5 | 42.17 ± 2.49 | 23.75 ± 2.43 | 29.67 ± 1.56 | 26.12 ± 2.01 | 37.8 ± 1.20 | 29.25 ± 1.65 |
| 4 | 50.17 ± 2.86 | 30 ± 1.56 % | 37.11 ± 1.35 | 34.5 ± 2.33 | 43 ± 1.05 | 36.25 ± 2.58 |
| 4.5 | 53.17 ± 1.87 | 37.62 ± 2.11 | 44.56 ± 1.79 | 43 ± 2.12 % | 47.6 ± 2.65 | 44.00 ± 0.09 |
| 5 | 59.00 ± 1.67 | 48.62 ± 1.56 | 54.33 ± 1.43 | 49.12 ± 1.21 | 51.2 ± 2.31 | 53.50 ± 2.69 |
| 5.5 | 63.00 ± 2.31 | 51.12 ± 1.21 | 61.78 ± 2.09 | 55.62 ± 2.61 | 59.6 ± 2.32 | 58.50 ± 1.33 |
| 6 | 66.33 ± 2.49 | 63.5 ± 2.39 | 68.78 ± 2.56 | 62.62 ± 3.32 | 63.8 ± 2.04 | 61.25 ± 1.12 |
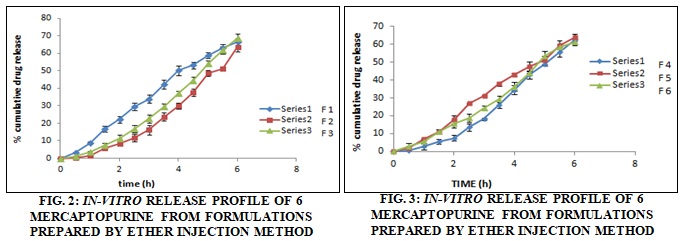
TABLE 4: RELEASE PROFILE OF FORMULATIONS PREPARED BY USING ETHER INJECTION METHOD USING VARIOUS MODELS
| Formulation | R2 | Korsmeyer | Best fit
model |
Release
mechanism |
||||
| Zero | First | Hixoncrowell | Higuchi | R2 | N | |||
| F 1 | 0.991 | 0.992 | 0.991 | 0.985 | 0.989 | 1.239 | First | Super case II |
| F 2 | 0.958 | 0.899 | 0.958 | 0.880 | 0.992 | 2.059 | korsmayer | Super case II |
| F 3 | 0.978 | 0.923 | 0.978 | 0.913 | 0.998 | 1.704 | Korsmayer | Super case II |
| F 4 | 0.970 | 0.931 | 0.970 | 0.900 | 0.997 | 1.824 | Korsmayer | Super case II |
| F 5 | 0.995 | 0.984 | 0.995 | 0.979 | 0.984 | 1.418 | Zero & Hixon | Super case II |
| F 6 | 0.984 | 0.949 | 0.984 | 0.932 | 0.994 | 1.257 | Korsmayer | Supercase II |
CONCLUSION: Anticancer drug delivery by using liposome is a new strategy with the potential to maximize the anticancer effect of a drug and reduce systemic toxicity. In this study, we have demonstrated the effectiveness of sustained delivery of the anticancer agent 6 Mercaptopurine using liposome as a drug delivery system, thus increasing bioavailability at cancer site and reduction of systemic toxicity.
A liposome is a safe, effective, homogeneous, injectable, and stable formulation for delivery of 6 Mercaptopurine, and this approach represents an attractive technology platform for the delivery of other clinically important anticancer drugs. The in-vivo studies may be done to further confirm the bioavailability of the drug and determine the drug release behavior inside the body.
ACKNOWLEDGEMENT: I am very much thankful to my guide Mr. Swatantra K. S. Kushwaha and Pranveer Singh Institute of Technology, Kanpur (UP) for providing best lab facilities necessary for the completion of the research project. I am highly obliged to Dabur Pharmaceuticals, Baddi for providing gift sample of 6 Mercaptopurine.
CONFLICT OF INTEREST: Nil
REFERENCES:
- Bangham AD, Glover JC, Hollingshead S and Pethica BA: Biochem J 1962; 84: 513.
- Chetanachan P, Akarachalanon P, Worawirunwong D, Dararutana P, Bangtrakulonoth A, Bunjop M and Kongmuang S: Advanced Materials Research 2008; 55(57): 709-71.
- Trommer H and Neubert RHH: Screening for new antioxidative compounds for topical administration using skin lipid model systems. J Pharm Pharmaceut Sci 2005; 8(3): 494-506.
- Kozubek A, Gubernator J, Przeworska E and Stasiuk M: Liposomal drug delivery, a novel approach: PLARosomes. Acta Biochimica Polonica 2000; 43(7): 639-649.
- Honarpisheh H, Rao MVSN and Mozafari MR: Micron 2007; 38: 804-818.
- Sharma VK, Mishra DN, Sharma AK and Srivastava B: IJCPR 2010; 1(2): 6-16.
- Lasic DD: Novel applications of liposomes. Tibtech 1998; 16: 307-321.
- Chrai SS, Murari R and Imran A: Liposomes: a review. Bio Pharm 2001; 14(11): 10-14.
- Andreas W and Karola VU: Liposome technology for industrial purposes. J Drug Deliv 2011; 2011: 9.
- Sharma VK, Mishra DN, Sharma AK and Srivastava B: IJCPR 2010; 1(2): 6- 16
- Hemanthkumar M and Spandana V: Liposomal encapsulation technology a novel drug delivery system designed for ayurvedic drug preparation. IRJP 2011; 2(10): 4-7.
- Mozafari MR: Liposomes: an overview of manufacturing techniques. Cellular & Molecular Biology Letters 2005; 10: 717-719.
- Barbeau J, Cammas-Marion S, Auvray P and Benvegnu T: J Drug Delivery 2011; 2011: 1-11.
- Anwekar H, Patel S and Singhai AK: Liposome- as drug carriers. Int J of Pharm & Life Sci (IJPLS) 2011; 2(7): 945- 951.
- Mozafari MR, Johnson C, Hatziantoniou S and Demetzos C: Nanoliposomes and their applications in food nanotechnology. J Liposome Research 2008; 18: 309-327.
How to cite this article:
Pateriya K, Kushwaha SK, Bijauliya RK and Chanchal DK: Liposome based sustained release delivery system for anticancer drug. Int J Life Sci & Rev 2018; 4(6): 98-03. doi: 10.13040/IJPSR.0975-8232.IJLSR.4(6).98-03.
All © 2015 are reserved by International Journal of Life Sciences and Review. This Journal licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License.
Article Information
2
98-103
633
1332
English
IJLSR
K. Pateriya *, S. K. Kushwaha, R. K. Bijauliya and D. K. Chanchal
Paramveer Singh Institute of Technology (PSIT), Kanpur, Uttar Pradesh, India.
pateriya7kriti@gmail.com
08 December, 2017
24 March, 2018
21 June, 2018
10.13040/IJPSR.0975-8232.IJLSR.4(6).98-03
30 June, 2018