


STUDY OF DISSOLUTION ENHANCEMENT OF POORLY WATER SOLUBLE GLIPIZIDE BY USING SOLID DISPERSION TECHNIQUE
HTML Full TextSTUDY OF DISSOLUTION ENHANCEMENT OF POORLY WATER SOLUBLE GLIPIZIDE BY USING SOLID DISPERSION TECHNIQUE
Md. Shozan Mondal *, Mehedi Hasan Emon, Sharif Md. Abuzar, Md. Ariful Islam, Md. Tauhidul Islam and Md. Ashraful Islam
Department of Pharmacy, University of Asia Pacific, Dhanmondi, Dhaka - 1209, Bangladesh.
ABSTRACT: The aim of the present research work is to design and evaluate controlled release transdermal patches of selected cardiovascular drug Atenolol. To optimize the prepared formulations by different trial approaches. To design and formulate transdermal patch of Atenolol by mercury substrate method using a few selected polymers viz., HPMC, PVP, EVA, Eudragit RS and RL100 in various proportions and Di-n-butyl Phthalate for the release retardant controlled release transdermal patch. Atenolol was chosen as a model drug for study since it possesses near ideal characteristic that a drug must have in formulating a drug delivery system such as low molecular weight, high lipid solubility, effective in low plasma concentration. It also means multiple administrations with subsequent lack of patient compliance.
Keywords: Glipizide, Polymers, Dissolution, Water Solubility, Solid Dispersion
INTRODUCTION: The progress in the treatment of diseases has been evident within an upsurge in the development of new drugs. An estimated 40% of these drugs are poorly water soluble. Although most of the drugs have encouraging experimental data obtained in-vitro, the in-vivo results have been disappointing. The attributes include
- Poor absorption, rapid degradation, and lamination (peptides and protein) resulting in insufficient concentration.
- Drug distribution to other tissues with high drug toxicities (anticancer drugs).
- Poor solubility of drugs and
- Fluctuations in plasma levels owing to unpredictable bioavailability.
- The enhancement of oral bioavailability of such poorly water-soluble drugs remains one of the most challenging aspects of drug development. The development of solid dispersions as a practically viable method to enhance the bioavailability of poorly water-soluble drugs overcame the limitations of previous approaches such as salt formation, solubilization by co-solvents, and particle size reduction 1.
Studies revealed that drugs in solid dispersion need not necessarily exist in the micronized state. A fraction of the drug might molecularly disperse in the matrix, thereby forming a solid dispersion 2. When the solid dispersion is exposed to aqueous media, the carrier dissolves and the drug releases as fine colloidal particles 3. The resulting enhanced surface area produces a higher dissolution rate and bioavailability of poorly water-soluble drugs. Also, in solid dispersions, a portion of drug dissolves immediately to saturate the gastrointestinal tract fluid and excess drug precipitates as fine colloidal particles or oily globules of submicron size 4.
The oral route of drug administration is the most common and preferred method of delivery due to convenience and ease of ingestion 5. From a patient’s perspective, swallowing a dosage form is a comfortable and familiar means of taking medication 6.
As a result, patient compliance and hence, drug treatment is typically more effective with orally administered medications as compared with other routes of administration, for example, parenteral. Although the oral route of administration is preferred, for many drugs, it can be a problematic and inefficient mode of delivery for several reasons 7. Limited drug absorption resulting in poor bioavailability is paramount amongst the potential problems that can be encountered when delivering an active agent via the oral route.
When delivering an active agent orally, it must first dissolve in gastric and intestinal fluids before it can then permeate the membranes of the GI tract to reach systemic circulation8. Therefore, a drug with poor aqueous solubility will typically exhibit dissolution rate limited absorption, and a drug with poor membrane permeability will typically exhibit permeation rate limited absorption9. Hence, two areas of pharmaceutical research that focus on improving the oral bioavailability of active agents include: (i) enhancing solubility and dissolution rate of poorly water-soluble drugs and (ii) enhancing the permeability of poorly permeable drugs 10.
This article focuses on the former, in particular, the use of solid dispersion technologies to improve the dissolution characteristics of poorly water-soluble drugs and in turn their oral bioavailability. Numerous solid dispersion systems have been demonstrated in the pharmaceutical literature to improve the dissolution properties of poorly water-soluble drugs. Other methods, such as salt formation, complexation with cyclodextrins, solubilization of drugs in solvent(s), and particle size reduction have also been utilized to improve the dissolution properties of poorly water-soluble drugs. However, there are substantial limitations with each of these techniques 11. On the other hand, the formulation of drugs as solid dispersions offers a variety of processing and excipients options that allow for flexibility when formulating oral delivery systems for poorly water-soluble drugs. Much of the research that has been reported on solid dispersion technologies involve drugs that are poorly water-soluble and highly permeable to biological membranes as with these drugs dissolution is the rate-limiting step to absorption12. Hence, the hypothesis has been that the rate of absorption in vivo will be concurrently accelerated with an increase in the rate of drug dissolution.
Future Prospects of Solid Dispersion: In spite of many advantages of solid dispersion, issues related to preparation, reproducibility, formulation, scale-up, and stability limited its use in commercial dosage forms for poorly water-soluble drugs. Successful developments of solid dispersion systems for preclinical, clinical and commercial use have been feasible in recent years due to the availability of surface-active and self-emulsifying carriers with relatively low melting points.
The preparation of dosage forms involves the dissolving of drugs in melted carriers and the filling of the hot solutions into gelatin capsules. Because of the simplicity of manufacturing and scale-up processes, the physicochemical properties and as expected to change significantly during the scale up. For this reason, the popularity of the solid dispersion systems to solve difficult bioavailability issues concerning poorly water-soluble drugs will grow up rapidly. Because of the dosage form can be developed and prepared using small amounts of drug substances in early stages of the drug development process, the system might have an advantage over such other commonly used bioavailability enhancement techniques as micronization of drugs and soft gelatin encapsulation.
One major focus of future research will be the identification of new surface-active carriers and self-emulsifying carriers for solid dispersion. Only a small number of such carriers are currently available for oral use. Some carriers that are used for topical application of drug only may be qualified for oral use by conducting appropriate toxicological testing. One limitation in the development of solid dispersion system may be the inadequate drug solubility in carriers so that a wider choice will increase the success of dosage form development. Research should also be directed toward the identification of vehicles or excipients that would retard or prevent crystallization of drugs from supersaturated systems. Attention should also be given to any physiological and pharmacological effects of carriers used. Many of the surface-active and self-emulsifying carriers are lipid in nature, so potential roles of such carriers on drug absorption, especially on their pglycoprotein-mediated drug efflux, will require careful consideration. In addition to bioavailability enhancement, much recent research on solid dispersion systems was directed towards the development of extended-release dosage forms.
It may be pointed out that this area of research has been reinvigorated by the availability of surface-active and self-emulsifying carriers and the development of new capsule filling processes. Because the formulation of solid dispersion for bioavailability enhancement and extended release of drugs may employ essentially similar processes, except for the use of slower dissolving carriers for the later use, it is expected that the research in these two areas will progress simultaneously and be complementary to each other.
MATERIALS AND METHODS:
Materials: Glipizide was a gift sample from Drug International Limited, Bangladesh. The other materials were obtained from different sources Table 1.
TABLE 1: MATERIALS USED IN THIS EXPERIMENT
| Name | Source | |
| Drug | Glipizide | Drug International Limited |
| Polymers | PEG 6000 | Loba Chemie, India |
| HPMC 6cps | Samsung, Korea | |
| Poloxamer 188 | BASF, Germany | |
| Poloxamer 407 | BASF, Germany |
Methods:
Preparation of dissolution medium, Simulated intestinal fluid [SIF, pH 6.8]: 6.8 gm monobasic Potassium Di-hydrogen phosphate was dissolved in 250 ml of water, and 770 ml of .2 N NaOH and 500 ml of water was added. The resulting solution was adjusted with either 0 .2N NaOH or 0 .2N HCl to a pH 6.8 ± 0.01 was diluted with water to 1000 ml.
TABLE 2: TYPES OF EQUIPMENT USED IN THIS EXPERIMENT
| Apparatus | Model |
| USP Type II Dissolution Apparatus | VEEGO, India |
| UV –VIS Spectrophotometer | UVmini-1240, Shimadzu corporation, Japan. |
| Sonicator (Power-Sonic 505) | HWASHIN TECHNOLOGY CO., Seoul, Korea |
| Electronic Balance | Shimadzu, BW 420 H. |
| pH Meter (pH 211 Microprocessor pH Meter) | HANNA Instruments, Romania |
| Vortex Meter (VM-2000) | DIGISYSTEM LABORATORY INSTRUMENTS INC. Taiwan |
| Sieve (Endecott’s Test Sieve) | Endecotts Limited, England |
TABLE 3: APPARATUS USED IN THIS EXPERIMENT
| Apparatus |
| Plastic Syringe (10ml) |
| Disposable Syringe filter |
| Desiccators (Silica) |
| Glass Vials (10ml) |
| Glass test tube |
Preparation of Solid Dispersion by Solvent Method: The solvent process either comprises dissolving a sparingly water-soluble drug and a water-soluble polymer, i.e. the carrier, in an organic solvent capable of dissolving both and removing the solvent by evaporation or comprises dissolving the drug in an organic solvent, dispersing the solution in the carrier and removing the solvent by evaporation to provide the desired solid dispersion.
At Table 4, 5, 6 (Formulation of Glipizide by solvent evaporation method with different polymer) – Glipizide was taken in the vial and methanol was added in each until Glipizide was completely dissolved. The drug was completely dissolved in the solvent. The polymer was added in the solution and sonicated it for 5 min. All solutions were dried by hot air. When the solutions were evaporated completely, they were stored in a desiccator. The formulations were withdrawn from vials, crushed in mortar and pestle, passed through 36-micron mesh. Then formulations were transferred in vials and stored in a desiccator.
TABLE 4: FORMULATION OF GLIPIZIDE SOLID DISPERSION BY CHANGING AMOUNTS OF POLOXAMER 188
| Formulation | SD | SD 2 | SD 3 |
| Glipizide | 300 mg | 500 mg | 700 mg |
| Poloxamer188 | 700 mg | 500 mg | 300 mg |
TABLE 5: FORMULATION OF GLIPIZIDE SOLID DISPERSION BY CHANGING AMOUNTS OF PEG6000
| Formulation | SD 4 | SD 5 | SD 6 |
| Glipizide | 300 mg | 500 mg | 700 mg |
| PEG 6000 | 700 mg | 500 mg | 300 mg |
TABLE 6: FORMULATION OF GLIPIZIDE SOLID DISPERSION BY CHANGING AMOUNTS OF HPMC (6cps) WITH PEG 6000
| Formulation | SD 7 | SD 8 | SD 9 |
| Glipizide | 700 mg | 700 mg | 700 mg |
| PEG 6000 | 300 mg | 300 mg | 0 |
| HPMC (6cps) | 0 | 1000 mg | 1000 mg |
Fusion Method: The melting or fusion method involves the preparation of the physical mixture of a drug and a water-soluble carrier and heating it directly until it melted. The melted mixture is then solidified rapidly in an ice-bath under vigorous stirring. The final solid mass is crushed, pulverized, and sieved. The drug was taken in a vial with polymer(s) and heated directly in paraffin bath within 80-90 ºC while stirring. When the mixture was turned into liquid, it was solidified directly in an ice bath. The final solid mass was crushed, pulverized, and sieved. Formulations of Table 7 and 8 which were used for fusion method.
TABLE 7: FORMULATION OF GLIPIZIDE SOLID DISPERSION BY CHANGING AMOUNTS OF POLOXAMER 188
| Formulation | SD 10 | SD 11 | SD 12 |
| Glipizide | 300 mg | 500 mg | 700 mg |
Poloxamer188 |
700 mg | 500 mg | 300 mg |
TABLE 8: FORMULATION OF GLIPIZIDE SOLID DISPERSION BY CHANGING AMOUNTS OF PEG 6000
| Formulation | SD 13 | SD 14 | SD 15 |
| Glipizide | 300 mg | 500 mg | 700 mg |
| PEG6000 | 700 mg | 500 mg | 300 mg |
Preparation of Standard Curve of Glipizide: To prepare a standard curve for Glipizide, 20 mg of Glipizide was accurately weighted & dissolved in 20 ml of Methanol in a 100 ml volumetric flask to produce a solution 1 mg/ml which was sonicated for 5 min. Then it was diluted up to 100 ml with dissolution medium and sonicated for another 10 min to make a stock solution. Then 1, 2, 3, 4, 5, 6, 7, 8, 9 & 10 ml of this solution was taken in 100 ml volumetric flask & 99, 98, 97, 96, 95, 94, 93, 92, 91 & 90 ml dissolution medium was respectively added to them for the purpose of serial dilution. This serial dilution was carried out to get different Glipizide concentration. These were then analyzed by UV spectrophotometer at 276 nm and absorbance values were noted. Then the absorbance values were plotted against drug concentration, and the standard curve of Glipizide was obtained.
TABLE 9: FORMULATION OF GLIPIZIDE SOLID DISPERSION BY CHANGING AMOUNTS OF POLOXAMER 188
| Formulation | SD 16 | SD 17 | SD 18 |
| Glipizide | 700 mg | 700 mg | 700 mg |
Poloxamer 407 |
300 mg | 300 mg | 0 mg |
HPMC 6cps |
0 mg | 1000 mg | 1000mg |
TABLE 10: ABSORBANCE VALUES OF THE STANDARD SOLUTION OF GLIPIZIDE FOR STANDARD CURVE USING DISTILLED WATER
| Concentration (µgm/ml) | Absorbance |
| 0 | 0 |
| 2 | 0.04 |
| 4 | 0.081 |
| 6 | 0.131 |
| 8 | 0.162 |
| 10 | 0.177 |
| 12 | 0.245 |
| 14 | 0.283 |
| 16 | 0.291 |
| 18 | 0.342 |
| 20 | 0.411 |
In-vitro Dissolution Study of Glipizide from Solid Dispersion: In-vitro dissolution study was performed in a paddle type Dissolution Apparatus (USP Type II) VEEGO, India. A fixed amount of solid dispersion containing 20 mg equivalent Glipizide from each batch was calculated for dissolution purpose. SIF (pH 6.8) was used as dissolution media. 900 ml of SIF (pH 6.8) was used as dissolution medium in each dissolution basket at a temperature of 37 °C and a paddle speed of 75 rpm. The fixed amount of solid dispersion from each batch was weighed and transferred in each dissolution basket. The dissolution was carried out for 1 hour, and 10 ml sample was withdrawn at predetermined intervals of 5, 10, 15, 20, 30, 40, 50 & 60 min. Every time 10 ml dissolution sample was compensated by another fresh 10 ml SIF (pH 6.8).
Dissolution samples were withdrawn with the help of the disposable syringe filter and were kept in a test tube. The dissolution samples were then analyzed spectrophotometrically in a UV-VIS spectrophotometer (UV-mini-1240, SHIMADZU CORP., and Kyoto, Japan). The dissolution study for each batch was performed in duplicate.
RESULTS AND DISCUSSION:
In-vitro Release of Glipizide with poloxamer 188 by using solvent evaporation method: In the dissolution study of Fig. 3 where poloxamer 188 was used in the solvent evaporation method, enhanced drug release was observed as compared to the pure drug. In this figure, three formulations were used to study the dissolution profile. Each formulation contains 300, 500, 700 mg of Glipizide and 700, 500, 300 mg of poloxamer were used which were indicated by SD1, SD2, SD3 respectively. The dissolution profile was increased concerning the % release drug. The % release of SD1, SD2, SD3 were 75.91, 75.96 and 88 % respectively after 1 h. The % release value for pure Glipizide was found approximately 22.69%. So the best result was observed solid dispersion SD3 (88 %) release of Glipizide in-vitro test. SD3 formulation contained 700 mg of Glipizide and 300 mg of poloxamer 188.
In-vitro Release of Glipizide with PEG 6000 by using Solvent Evaporation Method: In the dissolution study of Fig. 5 where PEG 6000 was used in the solvent evaporation method, enhanced drug release was observed as compared to the pure drug. In this figure, three formulations were used to study the dissolution profile. Each formulation contains 300, 500, 700 mg of Glipizide and 700, 500, 300 mg of PEG 6000 were used which were indicated by SD4, SD5, SD6 respectively. The dissolution profile was increased concerning the pure drug. The % release of SD4, SD5, SD6 were 82.50, 83.40 and 86.55 % respectively after 1 hour. The % release value for pure Glipizide was found approximately 22.69%. So the best result was observed solid dispersion SD6 (86.55%) release of Glipizide in-vitro test. SD6 formulation contained 700 mg of Glipizide and 300 mg of PEG 6000.
In-vitro Release of Glipizide PEG 6000 and HPMC 6cps by using Solvent Evaporation Method: In the dissolution study of Fig. 7 where PEG 6000 and HPMC 6cps was used in the solvent evaporation method, enhanced drug release was observed as compared to the pure drug. In this figure, three formulations were used to study the dissolution profile. Each formulation contains 700 mg of Glipizide and 300, 300, 0 mg of PEG 6000 and 0, 1000, 1000 mg of HPMC 6cps were used which were indicated by SD7, SD8, SD9 respectively. The dissolution profile was increased concerning the pure drug.
The % release of SD7, SD8, SD9 were 53.68, 56.16 and 81.20% respectively after 1 h. The % release value for pure Glipizide was found approximately 22.69%. So, the best result was observed solid dispersion SD9 (81.20 %) release of Glipizide in vitro test. SD9 formulation contained 700 mg of Glipizide and 1000 mg of HPMC 6cps.
In-vitro Release of Glipizide using Poloxamer 407, HPMC 6cps by Solvent Evaporation Method: In the dissolution study of Fig. 9 where Poloxamer 407 and HPMC 6cps were used in solvent evaporation method, enhanced drug release was observed as compared to the pure drug. In this figure, three formulations were used to study the dissolution profile. Each formulation contains 700 mg of Glipizide and 300, 300, 0 mg of Poloxamer 407 and 0, 1000, 1000 mg of HPMC 6cps were used which were indicated by SD16, SD17, SD18 respectively.
The dissolution profile was increased concerning pure drug. The % release of SD16, SD17, SD18 were 95, 81, and 78 % respectively after 1 hour. The % release value for pure Glipizide was found approximately 22.69%. So the best result was observed solid dispersion SD16 (95%) release of Glipizide in vitro test. SD16 formulation contained 700 mg of Glipizide and 300 mg of Poloxamer 407.
In-vitro Release of Glipizide with Poloxamer 188 by using in the Fusion Method: In the dissolution study of Fig. 11 where poloxamer 188 was used in Fusion method, enhanced drug release was observed as compared to the pure drug. In this figure, three formulations were used to study the dissolution profile. Each formulation contains 300, 500, 700 mg of Glipizide and 700, 500, 300 mg of poloxamer 188 were used which were indicated by SD10, SD11, SD12 respectively. The dissolution profile was increased concerning pure drug. The % release of SD10, SD11, SD12 were 77.28, 66.05 and 52 % respectively after 1 h. The % release value for pure Glipizide was found approximately 22.69%. So the best result was observed solid dispersion SD10 (77.28 %) release of Glipizide in vitro test. SD10 formulation contained 300 mg of Glipizide and 700 mg poloxamer 188.
In-vitro Release of Glipizide with PEG 6000 by using in the Fusion Method: In the dissolution study of where PEG 6000 was used in Fusion method, enhanced drug release was observed as compared to the pure drug. In this figure, three formulations were used to study the dissolution profile. Each formulation contains 300, 500, 700 mg of Glipizide and 700, 500, 300 mg of PEG 6000 were used which were indicated by SD13, SD14, SD15 respectively. The dissolution profile was increased concerning the pure drug. The % release of SD13, SD14, SD15 were 79.97, 53.83 and 68.46 % respectively after 1 hour. The % release value for pure Glipizide was found approximately 22.69%. So, the best result was observed solid dispersion SD13 (79.97%) release of Glipizide in-vitro test. SD13 formulation contained 300 mg of Glipizide and 700 mg of PEG 6000.
Comparison between Solvent Evaporation and Fusion Method by using a Different Formulation of Glipizide and Poloxamer 188: In Fig.15 we can see that 45% and 40% release was obtained at 5 min from both solvent evaporation and fusion method and 5% at pure drug and a gradual increase occurred at 10 min, after that, after 60 min 65% release was obtained from fusion method and 75% release was obtained from solvent evaporation method where 23% release at pure drug. In Fig. 16 we can see that from solvent evaporation and fusion method % release is 79.97% and 53.54% at same formulation respectively in 60 min but in Fig. 17 releases is 85.55% and 63.83% from both solvent evaporation and fusion method respectively. So we can say that the higher dissolution rate was obtained from the solvent evaporation method.
Comparison between Solvent Evaporation and Fusion Method by using a Different Formulation of Glipizide and PEG 6000: In Fig. 18 we can see that 55% and 45% release was obtained at 5 min from both solvent evaporation and fusion method but a rare increment at 15 min at fusion method. After that, at 60 min 73% release was obtained from the fusion method and 85.12% release was obtained from the solvent evaporation method.
In Fig. 19 we can see that from solvent evaporation and fusion method % release is 83.53% and 53.54% at same formulation respectively but in Fig. 20 releases is 86.55% and 66.83% from both solvent evaporation and fusion method respectively at 60 min. So we can say that higher dissolution rate was obtained from the solvent evaporation method.

FIG. 1: PREPARED FORMULATIONS
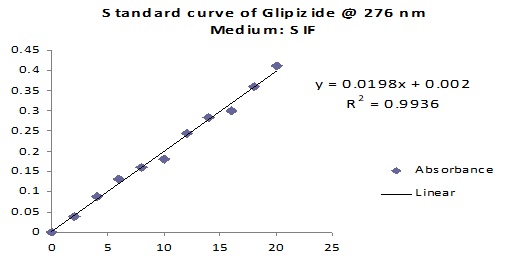
FIG. 2: STANDARD CURVE OF GLIPIZIDE
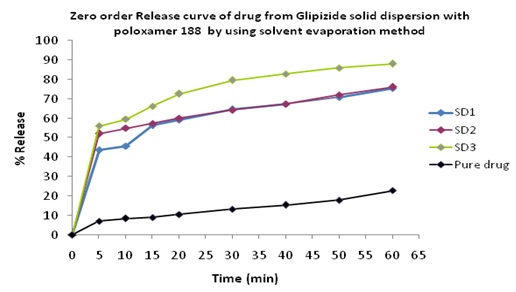
FIG. 3: EFFECT OF POLOXAMER 188 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION

FIG. 4: EFFECT OF POLOXAMER 188 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
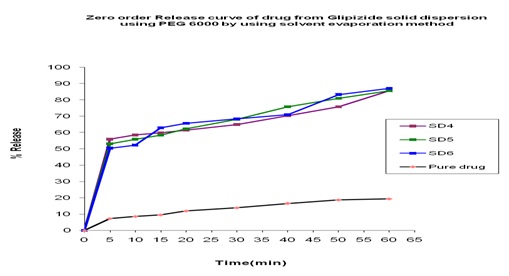
FIG. 5: EFFECT OF PEG 6000 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
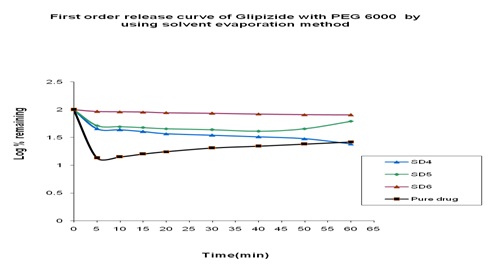
FIG. 6: EFFECT OF PEG 6000 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
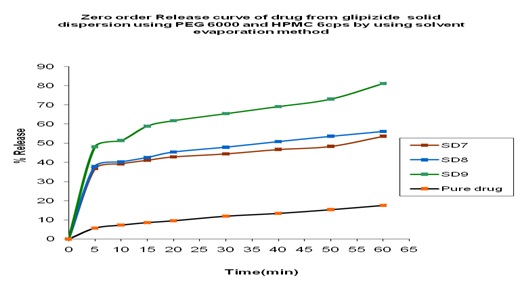
FIG. 7: EFFECT OF PEG 6000 AND HPMC 6cps ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
FIG. 8: EFFECT OF PEG 6000 AND HPMC 6cps ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
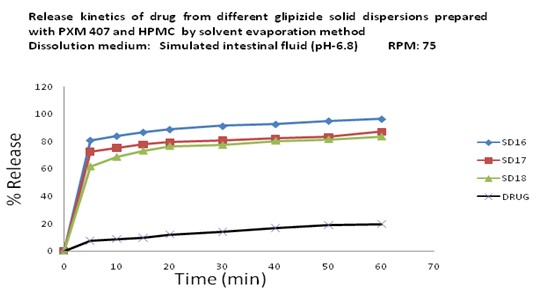
FIG. 9: EFFECT OF POLOXAMER 407 AND HPMC 6cps ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION

FIG. 10: EFFECT OF POLOXAMER 407 AND HPMC 6cps ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
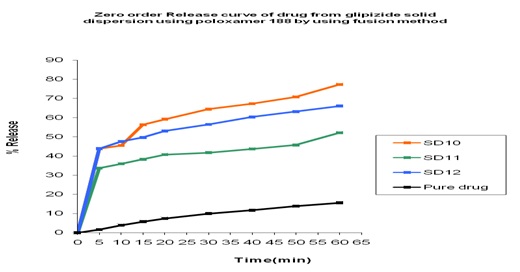
FIG. 11: EFFECT OF POLOXAMER 188 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
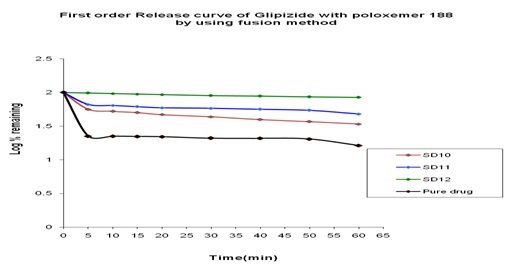
FIG. 12: EFFECT OF POLOXAMER 188 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
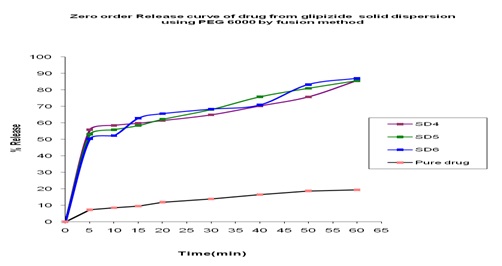
FIG. 13: EFFECT OF PEG 6000 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
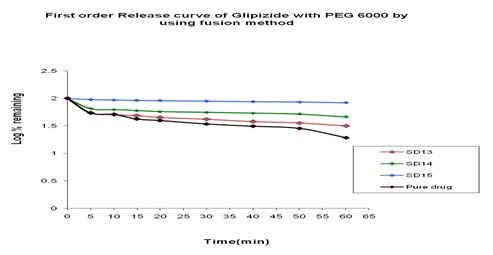
FIG. 14: EFFECT OF PEG 6000 ON THE DISSOLUTION PROFILE OF GLIPIZIDE SOLID DISPERSION
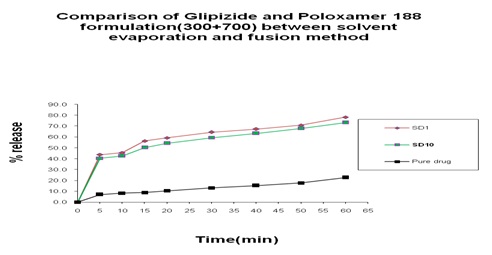
FIG. 15: COMPARISON OF GLIPIZIDE AND POLOXAMER 188 FORMULATION (300+700) BETWEEN SOLVENT EVAPORATION AND FUSION METHOD
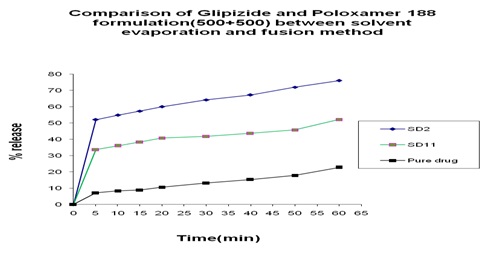
FIG. 16: COMPARISON OF GLIPIZIDE AND POLOXAMER 188 FORMULATION (500+500) BETWEEN SOLVENT EVAPORATION AND FUSION METHOD
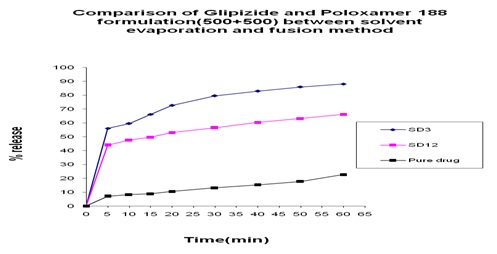
FIG. 17: COMPARISON OF GLIPIZIDE AND POLOXAMER 188 FORMULATION (700+300) BETWEEN SOLVENT EVAPORATION AND FUSION METHOD
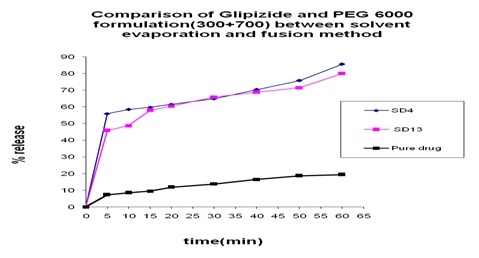
FIG. 18: COMPARISON OF GLIPIZIDE AND PEG 6000 FORMULATION (300+700) BETWEEN SOLVENT EVAPORATION AND FUSION METHOD
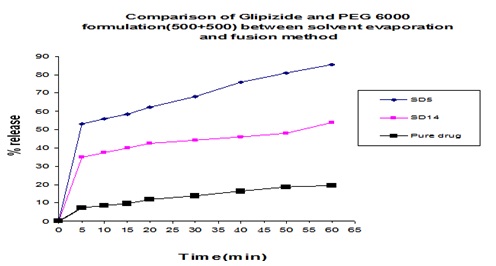
FIG. 19: COMPARISON OF GLIPIZIDE AND PEG 6000 FORMULATION (300+700) BETWEEN SOLVENT EVAPORATION AND FUSION METHOD
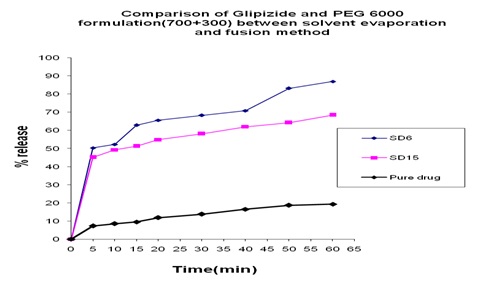
FIG. 20: COMPARISON OF GLIPIZIDE AND PEG 6000 FORMULATION (300+700) BETWEEN SOLVENT EVAPORATION AND FUSION METHOD
ACKNOWLEDGEMENT: The authors are indebted to Drug International Limited, Bangladesh and the University of Asia Pacific for necessary supports.
CONFLICT OF INTEREST: Nil
REFERENCES:
- Chiou WL and Riegelman S: Preparation and dissolution characteristics of several fast-release solid dispersions of griseofulvin. J Pharm Sci 1969; 58(12): 1505-10.
- Bayomi MA, Abanumay KA, and Al-Angary AA: Effect of inclusion complexation with cyclodextrins on the photostability of nifedipine in the solid state. Int J Pharm 2002; 243(1-2): 107-17.
- Betageri GV and Makarla KR Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int J Pharm 1995; 126: 155-60.
- Sekiguchi K and Obi N: Studies on the absorption of eutectic mixtures. I. A comparison of the behavior of eutectic mixtures of sulphathiazole and that of ordinary sulphathiazole in man. Chem Pharm Bull 1961; 9; 866-872.
- Goldberg A, Gibaldi M and Kanig L: Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures II experimental evaluation of a eutectic mixture: urea-acetaminophen system. J Pharm Sci 1966; 55; 482-87.
- Goldberg H, Gibaldi M and Kanig L: Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures I - theoretical considerations and discussion of the literature. J Pharm Sci 1965; 54; 1145-48.
- Hume-Rotherly W and Raynor GV: The Structure of Metals and Alloys,” Institute of Metals, London,
- Reed-Hill RE: Physical Metallurgy Principles. Van-Nostrand, Princetown, NJ,
- Greenhalgh DJ, Williams AC, Timmins P and York P: Solubility parameters as predictors of miscibility in solid dispersions. J Pharm Sci 1999; 88(11): 1182-90.
- Goldberg A, Gibaldi M and Kanig JL: Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures III - an experimental evaluation of griseofulvin- succinic acid solid solution. J Pharm Sci 1966; 55; 487-92.
- Chokshi R and Hossein Z: Hot–melt extrusion technique: a review. Int J Pharm Res 2004; 3: 3-16.
- Deitzel JM, Kleinmeyer J, Harris D and Tan NCB: The effect of processing variables on the morphology of electrospun nanofibers and textiles. Polym 2001; 42: 261-72.
How to cite this article:
Mondal MS, Emon MH, Abuzar SM, Islam MA, Islam MT and Islam MA: Study of dissolution enhancement of poorly water soluble glipizide by using solid dispersion technique. Int J Life Sci & Rev 2015; 1(5): 175-88. doi: 10.13040/IJPSR.0975-8232.IJLSR.1(5).175-88.
All © 2015 are reserved by International Journal of Life Sciences and Review. This Journal licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License.
Article Information
4
175-188
1121
2097
English
IJP
M. S. Mondal *, M. H. Emon, Sharif M. Abuzar, M. A. Islam, M. T. Islam and M. A. Islam
Department of Pharmacy, University of Asia Pacific, Dhanmondi, Dhaka, Bangladesh.
shozan_uap15@yahoo.com
23 February 2015
29 March 2015
28 April 2015
10.13040/IJPSR.0975-8232.IJLSR.1(5).175-88
01 May 2015